QUÍMICA ORGANOMETÁLICA. APLICACIONES EN CATÁLISIS UCM 2018 – 2019

ISABEL COLOMA MANJÓN – CABEZA
DAWID KRYSTIAN FELER BRYK
PATRICIA IZQUIERDO GARCÍA
ANA MARÍA LOBO OJEDA
RAMÓN LORTZING MARTÍNEZ
MARCOS MARTÍNEZ FERNÁNDEZ
ÍNDICE
1. INTRODUCCIÓN
1.1. PRECEDENTE HISTÓRICO
1.2. LA EVOLUCIÓN DE LAS INVESTIGACIONES DEL PALADIO
2. REACCIÓN DE HECK
3. CICLO CATALÍTICO
4. APLICACIONES
5. VENTAJAS
6.PÓSTER
6.PÓSTER
7. BIBLIOGRAFÍA
1. INTRODUCCIÓN
El descubrimiento del paladio por el químico inglés William Wollaston data de 1803.
Aunque fue Percival Norton Johnson, fundador de “Johnson Matthey”, una refinería de oro,
quien advirtió del uso del paladio en química. No es hasta 1946 cuando comienza a utilizarse
como catalizador para reacciones de hidrogenación de productos insaturados, debido a sus
propiedades alquenófilas. Sin embargo, hay que esperar hasta finales de los años 60 a que
Richard F. Heck demuestre el gran potencial del paladio como metal catalítico para
reacciones de acoplamiento C – C, y consolide su protagonismo dentro de la síntesis
orgánica moderna. De hecho, a causa del desarrollo de un campo de la química tan
importante como las metodologías de acoplamiento en química orgánica, Richard F. Heck,
E. Negishi y A. Suzuki fueron galardonados con el Premio Nobel de Química en 2010.
«Por el acoplamiento cruzado catalizado por paladio en síntesis
orgánica»
Las reacciones de acoplamiento son una herramienta fundamental en química
orgánica, ya que, a través de ellas se pueden generar enlaces C – C y C – X
(X = heteroátomo), con excelentes rendimientos y selectividades. Es por ello que han
llegado a adquirir un papel fundamental en la industria de productos fitosanitarios y
farmacéuticos. En esta última, en el año 2011, las reacciones de formación de enlaces
C – C supusieron el 11,5 % del total de procesos llevados a cabo para la síntesis de productos
de alto valor añadido, donde el 40,6 % de estas transformaciones se llevaron a cabo a través
de reacciones de tipo Suzuki-Miyaura o Mizoroki-Heck (1).
1.1.PRECEDENTE HISTÓRICO.
Wurtz (2) dio a luz al primer trabajo de acoplamientos C – C, naciendo así un nuevo
campo en química que no dejará de desarrollarse a partir de este momento. Persuadidos por
la aportación de Wurtz en esta incipiente rama química, Fittig y Tollens (3) completaron su
trabajo, entre 1862 y 1864, con el homoacoplamiento de haluros de arilo. A finales de esa
década, en 1869, Glaser (4) publicó un estudio donde describía el primer acoplamiento
Csp – Csp. En 1901 Ullmann (5) amplió el campo de utilidad del Cu, demostrando la
posibilidad de llevar a cabo acoplamientos de tipo Csp
2 –Csp
2. A pesar de estos remarcables
logros, estas nuevas aportaciones seguían estando limitadas en dos puntos clave:
a) El uso de metales en cantidades estequiométricas o superestequiométricas,
considerando que, además, éstos son poco solubles en el medio de reacción.
b) Los problemas de selectividad debido a las numerosas reacciones secundarias
observadas en este tipo de procesos, al llevar a cabo reacciones de acoplamiento
cruzado
Los trabajos de Job (en 1923) (6) y Meerwein (en 1939) (7) representan las primeras
demostraciones de que la formación de enlaces C – C es posible con el uso de metales en
cantidades catalíticas.
En la década de los años 70, Kochi demostraría la actividad catalítica de metales como
la plata (8), el cobre (9) y el hierro (10) en reacciones de acoplamiento, así como el estudio
mecanístico de estos procesos, consolidando las bases del actual entendimiento de estas
transformaciones.
A partir del final de la 2ª Guerra Mundial, la industria europea comienza a mostrar
cierto interés en el paladio, debido a la afinidad que mostraba por los dobles y triples
enlaces, al ser usado como catalizador en reacciones de hidrogenación de este tipo de
sustratos insaturados (11). Tras La Guerra, Europa debía ser reconstruida, y requería de
fuentes baratas de plásticos, así como de productos químicos de alto valor añadido
Dentro de este contexto surge, de forma algo accidental, el boom de “la química del
paladio” en la década de los años 50. En el instituto central de investigación Wacker
Chemie, químicos liderados por Hafner, comenzaron a trabajar en la síntesis de óxido de
etileno a partir de etileno. Al hacer pasar una corriente de etileno y oxígeno sobre un lecho
de carbón con paladio, se generó acetaldehído de forma inesperada como producto
mayoritario. Esta fructífera observación derivo en lo que ahora es conocido como el proceso
Wacker (12) [Esquema 1-1].

La investigación de Wacker Chemie sienta unas bases sólidas que permitirá el
desarrollo de la química organometálica del paladio en aplicaciones catalíticas de síntesis
orgánica durante todo lo que queda del siglo XX, y que, en la actualidad, sigue en marcha.
1.2.LA EVOLUCIÓN DE LAS INVESTIGACIONES DEL PALADIO.
Richard F. Heck continúa con sus estudios de postdoctorado en la compañía “Hércules
Powder Co.”. Heck planteó un experimento que permitió despejar las dudas: en acetonitrilo
a 0 ºC bajo una corriente de etileno, añadió acetato de fenilmercurio, observando una
transformación química inmediata (14). Esta observación culminó en 1968 con la
publicación de 7 artículos en un mismo año con Heck como único autor, sobre reacciones
de organomercuriatos con alquenos, en presencia de Li2[PdCl4] (15).
En 1968, de forma independiente y prácticamente a la par, Heck (16) y Mizoroki (17)
describieron el acoplamiento de haluros de arilo, bencilo y estirilo con alquenos utilizando
Pd como catalizador.
En 1975, tres grupos desarrollan prácticamente en paralelo la catálisis de reacciones
de acoplamiento cruzado de haluros de vinilo o arilo con acetilenos terminales: los de Heck
(18), Cassar (19) y Sonogashira (20). Sonogashira describió un método muy selectivo en el
que las transformaciones transcurren en condiciones suaves (a temperatura ambiente), al
usar Cu como co-catalizador.
En 1976, Negishi describió reacciones entre ioduros de arilo y acetiluros de zinc
catalizadas con [PdCl2(PPh3)2] de forma satisfactoria, al obtener predominantemente
alquinos derivados del proceso de acoplamiento (21). Dos años después, la síntesis de
cetonas a través del acoplamiento de cloruros de aroílo con organometálicos de estaño (22).
En 1979, Suzuki demostró que era posible llevar a cabo reacciones de acoplamiento
con participación de ácidos borónicos cuando se utilizaban cantidades estequiométricas de
Pd para catalizar los procesos (23).
Actualmente, los estudios se centran en el desarrollo profundo y el ajuste fino de cada
tipo de acoplamiento, a través de la variación de ligandos, la búsqueda de nuevos medios
de reacción alternativos a los disolventes tradicionales y la ampliación máxima posible del
tipo de sustratos y grupos funcionales compatibles con estos procesos de acoplamiento.
2. REACCIÓN DE HECK
La adición oxidativa de Pd (0) a un haluro, acetato o triflato insaturado, seguida de
una reacción con una molécula orgánica insaturada (generalmente un alqueno), produce
como reacción global la sustitución de un átomo de hidrógeno vinílico (o alílico) del grupo
insaturado [Esquema 2-1].

➢ Si hay dos hidrógenos en beta, la eliminación sin conduce
preferentemente al alqueno E.
➢ La reacción solo da rendimientos aceptables con Pd.
➢ Presiones elevadas favorecen la velocidad del proceso.
➢ La primera etapa esta desfavorecida en presencia de grupos
electrodadores.
➢ Los ioduros reaccionan mejor que los bromuros, y los cloruros
generalmente no reaccionan.
➢ La etapa de inserción está favorecida por sustituyentes
electroatractores en el alqueno.
Una investigación electroquímica para este clásico ciclo catalítico de la reacción de
Heck por Amatore y col. (24) enfatiza el papel mecanístico de los aniones acetato, haluro y
triflato. De acuerdo con sus investigaciones, la participación de complejos de paladio se
lleva a cabo en forma aniónica preferentemente a la neutra. De esta manera, la influencia
de los aniones en la velocidad de la adición oxidativa es justificable.
En el proceso de carbopaladiación, se lleva a cabo un control exquisito de la
quimio-, regio- y estereoselectividad (25). Previamente a la inserción la molécula orgánica
insaturada, se produce la coordinación de esta al paladio vía polar (P) o no-polar (N), que
se considera un proceso concertado (26) [Figura 2-2]:

La polaridad del medio, la naturaleza del ligando y del anión rigen si el proceso va
vía P o N. En definitiva, la reacción de Heck está gobernada por la densidad electrónica y
el impedimento estérico de la molécula orgánica insaturada.
Un estudio de la arilación del estireno (27), llevado a cabo en condiciones
estequiométricas, mostró que la regioselectividad dependía del grado de carácter catiónico
del intermedio aril-paladio. Las condiciones de reacción que promueven la vía P
disminuyen la regioselectividad de la reacción. La flexibilidad conformacional del ligando
quelante juega también un papel clave en el proceso [Figura 2-2].

Las posibles rutas de β-eliminación de hidruro otorgan problemas mayores de
regioselectividad, así como la posibilidad de que la molécula orgánica insaturada se
reinserte al complejo de paladio, produciendo productos de isomerización.
A pesar de estas dificultades, se han desarrollado variantes enantioselectivas de la
reacción de Heck (28). Ligandos quelantes quirales (29) permiten inducir la quiralidad en
el producto como, por ejemplo, (R)-BINAP:

Mayor enantioselectividad puede conseguirse cuando las condiciones de reacción
favorecen la vía P, ya que el ligando quiral permanece unido al paladio de forma bidentada
y la inducción quiral es más efectiva.
3. CICLO CATALÍTICO
El ciclo catalítico de la reacción de Heck es el que se observa en el Esquema (3-1).
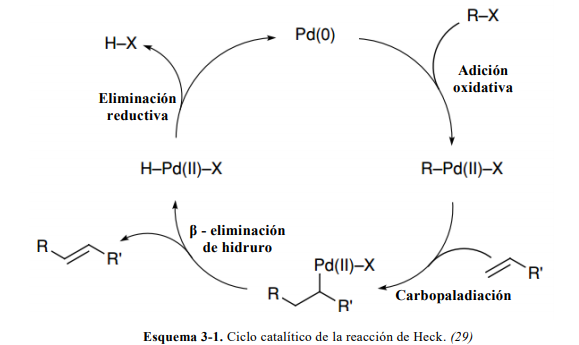
El catalizador
El objetivo último del ciclo catalítico es la formación de un enlace C – C de
acoplamiento cruzado. Los precursores catalíticos más habituales en la reacción de Heck
son de paladio con estado de oxidación formal (II), como por ejemplo [Pd(OAc) 2], y se
encuentran en equilibrio con compuestos de Pd(0). Este equilibrio se favorece si en el medio
de reacción hay ligandos neutros dadores de pares de electrones como la trifenilfosfina
(PPh3) (30).

La formación del complejo de paladio catalíticamente activo, [Pd(PPh3)2] será la etapa
determinante para que pueda iniciarse el ciclo. Una vez formado el complejo de Pd(0) tiene
lugar la adición oxidativa. (30)
Adición oxidativa
El enlace R – X se puede romper fácilmente para formar los enlaces R – [Pd] – X. La
velocidad con que a la que esta etapa tenga lugar también depende de las propiedades
químicas de X, la velocidad varía de la siguiente forma: I > OTf > Br > Cl. (31)
Carbopaladiación
Inicialmente, la olefina se asocia al complejo de paladio, lo que requiere de la
disociación de un ligando existente. El ciclo produce un enlace fuerte Pd – X y un enlace
débil Pd – PR3. Por ello, se producirá la salida de un ligando fosfina y la olefina se asociará
al entorno plano cuadrado del paladio, como se observa en la ruta A (X = Cl, Br, I). Este
mecanismo inicialmente propuesto, resulta ser poco realista, ya que el compuesto que
planteado como intermedio no presenta propiedades catalíticas (32). Esta situación llevó a
la introducción de triflatos o acetatos (X), ligandos que se asocian poco al paladio, como
grupos salientes y que de esta manera se produzca la salida de X y no del ligando fosfina,
esta alternativa se muestra en la ruta B. (29)

Como se observa en los esquemas de ambas rutas, una vez que se produce la
asociación de la olefina, tiene lugar de forma irreversible la inserción de la olefina en el
enlace Pd – R (29), el grupo R ataca como nucleófilo a la olefina que, al estar coordinada a
un metal, en este caso el Pd, es un electrófilo. Si la olefina está sustituida, esta etapa será
clave en lo que respecta a la estereoquímica del producto final.
β-eliminación
La β-eliminación es una reacción de descomposición de un complejo metal-alquilo
para dar un complejo metal-hidruro y un alqueno. Siendo su reacción inversa a la inserción
A
B
10
1,2 de alquenos en enlaces M-H. En esta etapa se obtiene el producto de acoplamiento
cruzado C-C.

El requisito fundamental es la existencia de al menos un hidrógeno en posición beta.
También, es necesario que el compuesto tenga 16 electrones de valencia como máximo, ya
que el centro metálico ha de estar electrónica y coordinativamente insaturado. Además, se
debe permitir la coplanaridad del metal con dicho hidrógeno ya que se trata de un proceso
sin concertado (33).
El equilibrio inserción-eliminación se desplaza en el sentido de la inserción siempre
que existan grupos electroatrayentes en el alqueno, cuando el producto esté excesivamente
tensionado o grupos voluminosos impidan la disposición coplanaria del metal e hidrógeno
(33).
Eliminación reductora
La eliminación reductiva o reductora es la reacción inversa a la adición oxidativa.
Consiste en la formación de un compuesto A-B a partir de un complejo LnM(A)(B) en el
que A y B son ligandos sigma dadores:

Tal como indica su nombre, la reacción conlleva la reducción del estado de oxidación
del metal, volviendo a su valor original y reiniciando el ciclo catalítico. Experimentalmente
sea demostrado que se trata de una reacción intramolecular. Además, A y B deben de estar
en cis para poder dar lugar a esta (33)
4. APLICACIONES
Las reacciones de acoplamiento C – C presentan una gran variedad de aplicaciones,
ya que la generación de estos enlaces C – C, o bien C – X, son una herramienta muy
importante en química orgánica dado que permite construir esqueletos moleculares
complejos a partir de otros más sencillos (34). Concretamente, la reacción de Heck es una
de la que más aplicaciones tiene ya que se ha estudiado exhaustivamente, consiguiendo
mejoras en los catalizadores y, por tanto, en los rendimientos y selectividades.
Podemos encontrar las siguientes aplicaciones en función del tipo de industria,
encontrándose un mayor número en la farmacéutica.
1. Industria farmacéutica: gran impacto en este campo ya que a través de esta reacción
se han conseguido tratamientos contra diversas enfermedades.
- Tratamiento contra el SIDA: síntesis de la Rilpivirina. Se trata de un inhibidor
de la transcriptasa inversa

- Tratamiento contra el asma: síntesis de Montelukast, el cual bloquea los
receptores de los leucotrienos, inhibiendo de esta manera la broncoconstricción.

- Síntesis de la (-)-morfina: reacción en cadena que comienza con una reacción de
acoplamiento C – C. (34)

2. Industria textil: la reacción de Heck se utiliza para producir tintes de alta calidad.
(35)
3. Fabricación de diodos orgánicos para pantallas OLED. (35)
4. Síntesis de herbicidas: Prosulfuron

5. Aplicaciones en estudio: síntesis de (+)-discodermolida. Se trata de un compuesto
en estudio para futuros fármacos anticancerosos. (34)

5. VENTAJAS
La reacción de Mizoroki – Heck (1968-1973), para el acoplamiento de haluros de
arilo, bencilo y estirilio con alquenos, utilizando Pd como catalizador, ofrece múltiples
ventajas a nivel industrial y en los sectores farmacéutico, textil y agroalimentario:
• Es el mejor método para la preparación de olefinas disustituidas a partir de olefinas
monosustituidas.
• La cinética y el rendimiento de la reacción no se ven afectados, o lo hacen muy poco,
por la naturaleza electro – dadora o electro – aceptora de los sustratos.
• Por este motivo, la reacción es compatible con prácticamente cualquier grupo
funcional, aunque suele ir más rápido con olefinas que poseen grupos
electroatractores.
• El grado de sustitución de la olefina tiene una alta influencia sobre la cinética de
reacción, controlando este parámetro, es posible llevar a cabo acoplamientos
quimioselectivos.
• La estereoespecificidad de estos procesos —inserción migratoria de la olefina y
𝛽-eliminación del hidruro de Pd— es de tipo sin.
• No es sensible al aire ni a la humedad, y puede llevarse a cabo en una amplia gama
de disolventes, aunque es conveniente desgasificarlos para mantener la actividad del
catalizador de Pd.
Así, la principal ventaja de la reacción de Heck, y la característica fundamental que la
diferencia del resto de reacciones de acoplamiento C – C, es que no es necesario el empleo
de un compuesto organometálico como compañero de acoplamiento y, por tanto, no es
necesaria una etapa de transmetalación para que el proceso tenga lugar.
Esperamos que la lectura de nuestro trabajo haya sido de su agrado.
Atentamente: Isa, David, Patricia, Ana, Ramón y Marcos.
6. PÓSTER

Esperamos que la lectura de nuestro trabajo haya sido de su agrado.
Atentamente: Isa, David, Patricia, Ana, Ramón y Marcos.
6. PÓSTER

7. BIBLIOGRAFÍA
1. Roughley, S. D. 3451, 2011, A. M. Jordan, J. Med. Chem., Vol. 54.
2. A.Wurtz. 275, 1855, Ann. Chim. Phys., Vol. 44.
3. Fittig, R. 361, 1872, Justus Liebigs Ann. Chem., Vol. 121.
4. C. Glaser, Ber. 422, 1869, Dtsch. Chem. Ges., Vol. 2.
5. F. Ullmann, J. Bielecki. 2174, 1901, Ber. Dtsch. Chem. Ges., Vol. 34.
6. A. Job, R. Reich. 1439, 1923, C. R. Hebd. Seances Acad. Sci., Vol. 177.
7. H. Meerwein, E. Buchner, K. van Emster. 237, 1939, J. Prakt. Chem., Vol. 152.
8. J. K. Kochi, M. Tamura. 1483, 1971, J. Am. Chem. Soc., Vol. 83.
9. J. K. Kochi, M. Tamura. 1485, 1971, J. Am. Chem. Soc., Vol. 93.
10. J. K. Kochi, M. Tamura. 1487, 1971, J. Am. Chem. Soc., Vol. 93.
11. Mozingo, R. 77, 1946, Org. Synth., Vol. 26.
12. J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, H. Kojer, R. Rüttinger.
176, 1959, Angew. Chem., Vol. 71.
13. Tsuji, J., Nagashima, H., Nemoto, H. 137, s.l. : Org. Synth., 1990, Vol. 7.
14. Cobalt and Palladium Reagents in Organic Synthesis: The Beginning. Heck, R.
F. 2006, Synlett, Vol. 2855.
15. Heck, R. F. 5518, 1968, J. Am. Chem. Soc., Vol. 90.
16. H. A. Dieck, R. F. Heck. 1133, 1974, J. Am. Chem. Soc., Vol. 96.
17. T. Mizoroki, K. Mori, A. Ozaki. 1505, 1973, Bull. Chem. Soc. Jpn., Vol. 46.
18. H. A. Dieck, F. R. Heck. 259, 1975, J. Organomet. Chem., Vol. 93.
19. Cassar, L. 253, 1975, J. Organomet. Chem., Vol. 93.
20. K. Sonogashira, Y. Tohda, N. Hagihara. 4467, 1975, Tetrahedron Lett., Vol. 16.
20. K. Sonogashira, Y. Tohda, N. Hagihara. 4467, 1975, Tetrahedron Lett., Vol. 16.
21. Negishi, E. 340, 1982, Acc. Chem. Res., Vol. 15.
22. D. Milstein, J. K. Stille. 3636, 1978, J. Am. Chem. Soc., Vol. 100.
15
23. H. A. Dieck, R. F. Heck. 1083, 1975, J. Org. Chem., Vol. 40.
24. Amatore, C. 33, s.l. : Acc. Chem. Res. , 2000. 314.
25. Beletskaya, I.P. 100, s.l. : Chem. Rev. , 2000. 3009.
26. Elschenbroich, C. Organometallics. Marburg, Germany : Wiley-VCH, 2006.
27. Akermark, B. 18, s.l. : Organometallics, 1999. 970.
28. Shibasaki, M. 576, s.l. : J. Organomet. Chem. , 1999. 1.
29. W. Carruthers, I. Coldham. Modern methods of organic synthesis. Cambridge :
Cambridge University Press, 2004.
30. UC Davis. Chemistry LibreTexts. Heck Reaction. [Última consulta: 30/12/2018.]
https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Supplemental_Modules_(In
organic_Chemistry)/Catalysis/Catalyst_Examples/Heck_Reaction.
31. The Heck reaction: mechanistic insight into a synthetically useful reaction.
Tambar, U. K. s.l. : Stoltz Group Literature Series, 2003.
32. Cabri, W., Candiani, I. 28, s.l. : Acc. Chem. Res., 1995. 2-7.
33. Astruc, Didier. Química Organometálica. Grenoble, Francia : Reverté, 2003.
978-3-540-46128-9.
34. Avedaño, M. C. Anales de la Real Academia Nacional de Farmacia (RANF). El
Premio Nobel en Química 2010. [Última consulta: 02/01/2019]
https://www.analesranf.com/index.php/aranf/article/viewFile/1142/1182 .
35. MSc Alejandro Víctor Martínez Esteban. Tesis Doctoral: Diseño de Catalizadores basados en Nanopartículas de Paladio, para Reacciones de Interés Sintético. iSQCH
No hay comentarios:
Publicar un comentario